Néhány évvel ezelőtt alakult az az önkéntes munkacsoport, amelynek célja az orvostechnikai eszközök nemzetközi szabályozásának minél gyorsabb ütemű világszintű harmonizációja. A Globális Harmonizációs Munkacsoport (GHTF, Global Harmonization Task Force) 2008-ban kezdte meg munkáját nemzeti orvostechnikai eszközszabályozó hatóságok, illetve az orvostechnikai eszközipar neves képviselőinek részvételével.
Napjainkban már igen jelentős létszámú a munkacsoport, többek közt a tagjai olyan nemzeti hatóságok, mint az US FDA, EU Bizottság, a Health Canada, a Japan PMDA, a China FDA, az ANVISA Brasil, az EUCOMED/EDMA, az ADVAMED, a JFMDA/JIRA, a COCIR/DITTA. A meghívott megfigyelők között található a WHO, az AHWP és az APEC is. A GHTF munkáját 2012-ben az Orvostechnikai eszközök nemzetközi szabályozóinak fóruma (IMDRF, International Medical Device Regulators Forum) vette át.
A munkacsoport a harmonizált orvostechnikai szabályozás érdekében a résztvevő hatóságokkal közösen egy olyan szabályozási keretrendszert dolgozott ki (GHTF UDI Guidance Document), amely helyi-, nemzeti- és globális szinten is alkalmazható. A dokumentumot 2013-ban frissítették, elérhető a következő linken:
http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-131209-udi-guidance.pdf
A dokumentum alapelveket tartalmaz az UDI rendszerre, az azonosításra, a címkére, az UDI adatbázisra és a speciális eszköztípusokra vonatkozóan. Fontos kiemelni a keretrendszerrel kapcsolatban, hogy a hatóságok által közösen elfogadott irányelveket tartalmazza, amelyek használata ugyanakkor nem kötelező. A nemzetközi tendenciák ismeretében elmondható, hogy több országban is elindult az IMDRF útmutatóval összhangban az orvostechnikai eszközök szabályozása.
Élen jár az US FDA szabályozás
Még 2013 szeptemberében publikálta az US FDA (Food and Drug Administration – az Egyesült Államok Élelmiszeripari és Egészségügyi Szervezete) azt az orvostechnikai eszközszabályozást, mely alapján az Egyesült Államokban forgalmazott legtöbb orvostechnikai eszköznek egyedi eszközazonosítót, UDI-t (unique device identification) kell hordoznia szemmel olvasható módon és vonalkódban egyaránt.
Az UDI rendszer segítségével az orvostechnikai eszközökkel kapcsolatos tevékenységek, események nyomon követése pontosabbá válik, vagyis az eszközökről az ellátási láncban forgó információk minősége javul, ami lehetővé teszi az FDA számára a termékkel kapcsolatos esetleges problémák gyorsabb észlelését, a célzott termékvisszahívást és ezáltal a magasabb szintű betegbiztonság elérését.
A szabályozás az eszközöket kockázati szintjük szerint 3 csoportba sorolja (Class I-III), amely meghatározza, hogy milyen adattartalommal és milyen határidővel kell azokra vonatkozóan az új rendszert bevezetni. A legelső határidő 2014. szeptember 24-e volt, ami a Class III típusú termékekre vonatkozott.
Az FDA szabályozás azokat a hazai gyártókat érinti, akik már most is forgalmaznak, vagy a jövőben tervezik az orvostechnikai eszközök forgalmazását az Amerikai Egyesült Államokban.
UDI rendszer
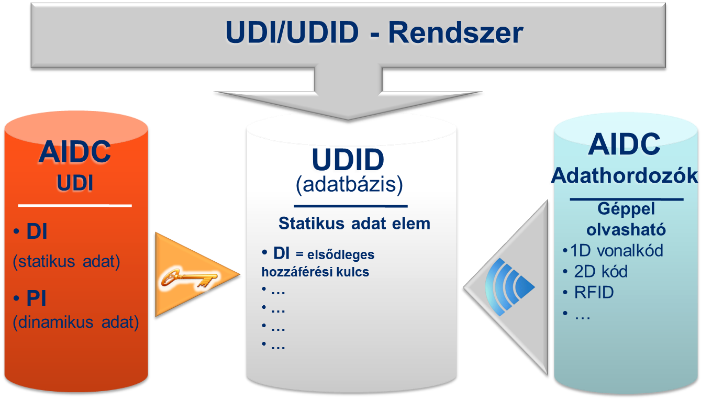
Az UDI (Unique Device Identification) Rendszer 3 fő elemből áll (1. ábra):
orvostechnikai eszközök azonosítása (UDI azonosítóval);
orvostechnikai eszközök jelölése (automatikus leolvasást lehetővé tevő eszközzel);
orvostechnikai eszközök törzsadatait tartalmazó adatbázis (UDID adatbázis).
A GS1 Magyarország (és a GS1 szervezet globális szinten) mind a három területen képes támogatni a partnereit, akiket már most érinthet az amerikai UDI szabályozás, ha orvostechnikai eszközöket exportálnak vagy tervezik az exportálást az Amerikai Egyesült Államokba.
Belátható időn belül az EU piacára is hasonló előírás várható, ami a hazai szereplőket egyaránt érinteni fogja, ezért is fontos az átjárható, harmonizált szabályozás a jogrendszerek között.
Tudjon meg többet az UDI szabályozásról a GS1 szabványrendszer egészségügyi megoldásairól! Jöjjön el a GS1 Globális Egészségügyi Konferenciájára 2015. október 20-22. között, Budapesten a Hotel Kempinski Corvinusba!